pDie strongPhenylketonurie (PKU) /strongist eine genetisch be-dingte Stoffwechselerkrankung, bei der durch eine Genmutation das Enzym Phenylalaninhydroxylase nicht in funktionstüchtiger Form produziert werden kann. Dadurch ist die normalerweise von der Phenyl-alaninhydroxylase katalysierte Umsetzung des mit der Nahrung aufgenommenen Phenylalanins in Ty-rosin nicht möglich, wodurch sich einerseits Phenyl-alanin im Körper anhäuft, während dem Organismus andererseits Tyrosin und die daraus erzeugten Pro-dukte fehlen (Stoffwechselblock). Das überschüssige Phenylalanin wird im Körper zu Phenylbrenztrauben-säure und anderen Produkten umgesetzt, die zu schweren Störungen der geistigen Entwicklung füh-ren. Die normalerweise aus Tyrosin erzeugten Stoff-wechselprodukte Melanin (dunkler Farbstoff in Haut und Haaren) und Thyroxin (Hormon, das u. a. eine bedeutende Rolle für die normale Entwicklung des heranwachsenden Körpers spielt) sind bei unbehan-delten PKU-Kranken kaum vorhanden, weshalb Albi-nismus (auffallend helle Haut und Haare) sowie Kre-tinismus (Kleinwüchsigkeit) zu den Symptomen die-ser Krankheit zählen. /p
pDie PKU ist eine der häufigsten genetisch bedingten Erkrankungen: etwa 1 von 8000 Neugeborenen ist davon betroffen. Je früher die Krankheit diagnosti-ziert werden kann, desto erfolgreicher können die sonst damit verbundenen Auswirkungen vermieden werden. Deshalb wird im Rahmen des emNeugebore-nenscreenings/em allen Säuglingen innerhalb der ersten drei Tage nach der Geburt Blut entnommen, um die Konzentration des darin enthaltenen Phenylalanins und Tyrosins zu bestimmen. /pp/ppDie nebenstehende Grafik zeigt das Ergebnis einer solchen Untersuchung. Die blau umrandeten Kästen markieren den Normbereich; die roten Punkte die im konkreten Fall gemessenen Werte./p
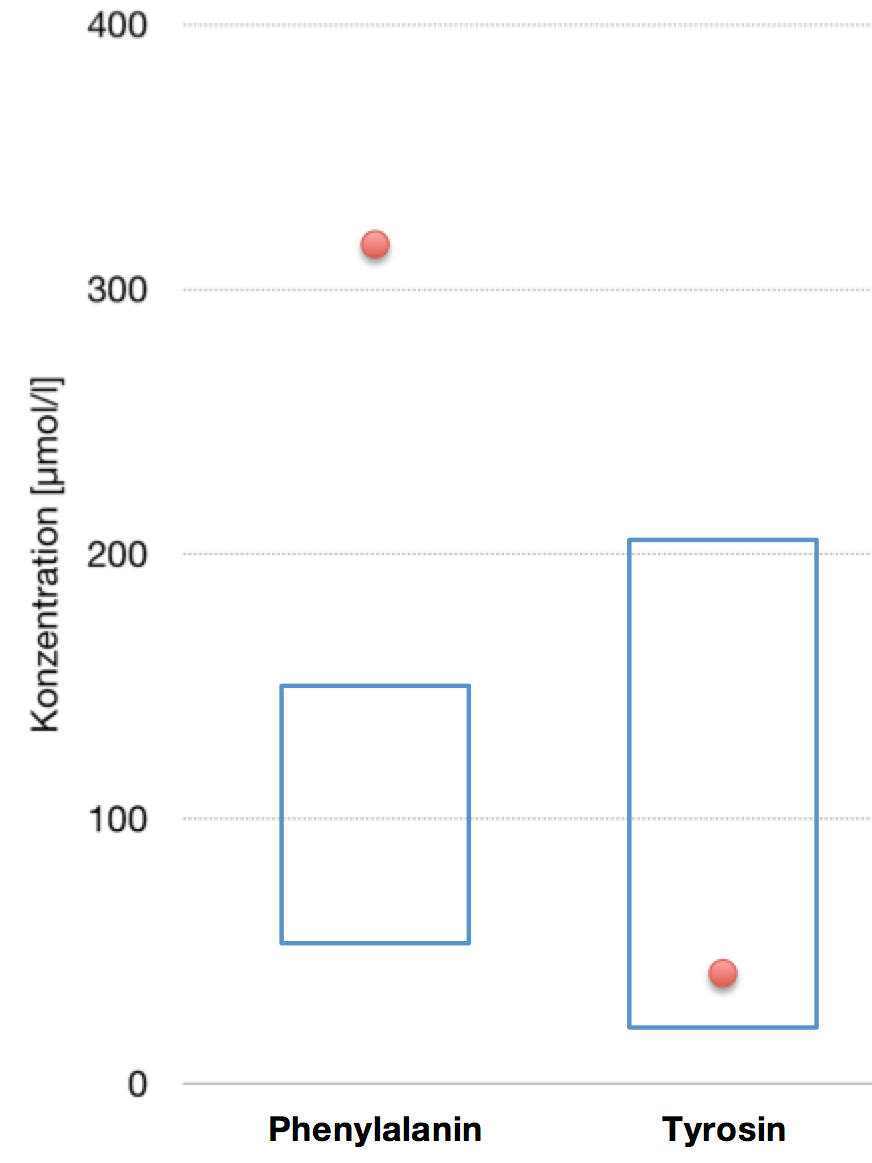
bei einem Neugeborenen
Pränataldiagnostik
pStoffwechselerkrankungen wie z. B. die PKU können mithilfe der strongPränataldiagnostik/strong (vorgeburtliche Dia-gnostik) auch bereits beim Ungeborenen festgestellt werden. Dazu benötigt man Zellen des Kindes, die man in diesem Fall durch eine Amniozentese ge-winnt. Da man hierbei mit einer Kanüle die Frucht-blase ansticht, um daraus Fruchtwasser (mit darin befindlichen Zellen des Kindes) zu entnehmen, ge-hört die Amniozentese – wie auch die Chorionzotten-biopsie oder die Nabelschnurpunktion – zu den eminva-siven/em Methoden der Pränataldiagnostik, wohingegen etwa Ultraschalluntersuchungen oder Blutuntersu-chungen bei der Mutter zu den emnicht-invasiven/em Me-thoden gezählt werden. Die Entscheidung, ob und welche Pränataldiagnostik eingesetzt wird, liegt bei den Eltern. /p

Heterozygotentest
pGenetisch bedingte Krankheiten (Erbkrankheiten) be-ruhen auf der Mutation eines Gens, das für ein be-stimmtes Protein, zumeist ein Enzym, codiert. Auf der Grundlage dieser mutierten Genvariante kann das betreffende Protein nicht in funktionstüchtiger Form produziert werden. Da alle Körperzellen jedoch einen diploiden Chromosomensatz und somit für jedes Gen emzwei/em Allele besitzen, ist die Chance sehr hoch, dass zumindest eines der Allele die intakte An-leitung zur Synthese des Proteins enthält. Das be-treffende Protein wird in dem Fall, dass eines der Al-lele mutiert, das andere Allel jedoch intakt ist, zwar nur in etwa halb so großer Menge hergestellt wie bei einer Person, die zwei intakte Allele besitzt, dennoch ist es auf jeden Fall vorhanden und aktiv, weswegen sich bei der betreffenden Person in der Regel keine Krankheitssymptome feststellen lassen. Diese treten nur auf, wenn embeide/em Allele des Gens mutiert sind und daher das codierte Protein emgar nicht/em in funktions-tüchtiger Form synthetisiert werden kann. /pp/ppWir können derartige Erbkrankheiten also als emdomi-nant-rezessive Erbgänge/em betrachten, bei denen die „Krankheit“ – d. h. das mutierte Allel – rezessiv (a) und die „Gesundheit“ – d. h. das intakte Allel – dominant (A) wirken. Zur phänotypischen Ausprä-gung der Krankheit kommt es nur im homozygot re-zessiven Fall (aa), während sich im homozygot domi-nanten (AA) sowie im heterozygoten Fall (Aa) die Gesundheit „durchsetzt“ und keine Krankheitssymp-tome zu beobachten sind./pp/ppPersonen, die phänotypisch gesund sind, können al-so die Genotypen AA oder Aa besitzen. Im homozy-goten Fall können sie auch stets nur ein dominantes „Gesundheits-Allel“ an ihre Kinder weitergeben; im heterozygoten Fall jedoch besteht eine 1:1-Chance, dass die Kinder dieser Person das „Gesundheits-“ bzw. das „Krankheits-Allel“ erben. Hat auch der Part-ner der betreffenden Person einen heterozygoten Genotyp, so wird sich statistisch gesehen bei einem Viertel der gemeinsamen Kinder der Genotyp aa (homozygot rezessiv) ergeben – diese Kinder wären dann auch phänotypisch krank./pp/ppUm bei Personen, die phänotypisch gesund sind, zu ermitteln, ob sie den Genotyp AA oder den Genotyp Aa besitzen, bedient man sich einer Methode, die als strongHeterozygotentest/strong bezeichnet wird. Hierbei misst man die Konzentration des Substrats bzw. des Pro-dukts des betreffenden Enzymproteins im Körper der Testperson und vergleicht das Ergebnis mit dem von Erkrankten und von anderen Gesunden./p
pDie Phenylketonurie (PKU) ist eine rezessiv vererbte Krankheit. Sie zeigt sich im Phänotyp, wenn bei-de Allele für das Enzym Phenylala-ninhydroxylase mutiert sind, so-dass im Körper keine funktions-tüchtige Phenylalaninhydroxylase synthetisiert werden kann. Fehlt dieses Enzym, kann das mit der Nahrung aufgenommene Phenyl-alanin nicht in Tyrosin umgesetzt werden. Um den PKU-Genotyp ei-nes phänotypisch gesunden Men-schen abzuleiten, wird dem Pro-banden eine hohe Dosis Phenyl-alanin verabreicht; anschließend wird ihm im Abstand von zwei Stunden Blut entnommen, und dessen Tyrosinkonzentration be-stimmt. Die Ergebnisse werden mit den Werten von PKU-Kranken und anderen Gesunden verglichen (siehe nebenstende Grafik)./p
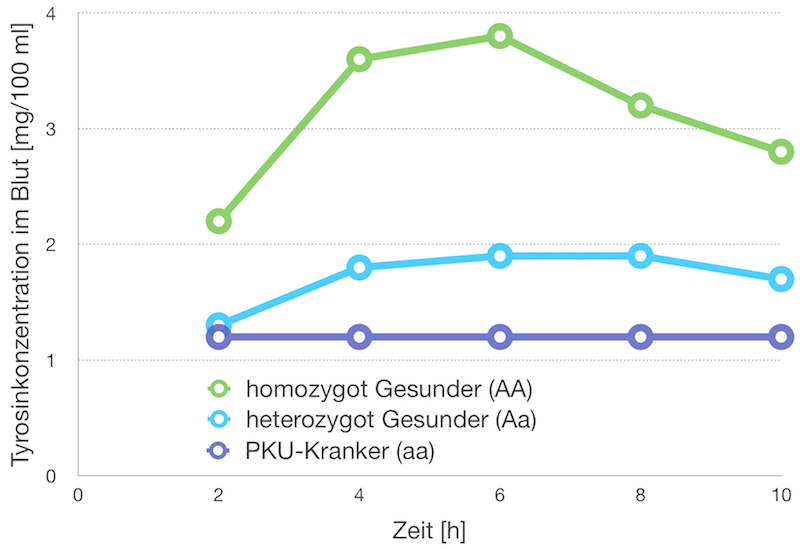
PKU-Kranke können Phenylalanin nicht in Tyrosin umwandeln; homo-zygot Gesunde setzen Phenylalanin schnell zu Tyrosin um; bei Hetero-zygoten ist dies aufgrund der geringeren Menge an Phenylalaninhy-droxylase nur langsam möglich.
Genetische Beratung
pEine stronghumangenetische Beratung/strong, oft auch als strongge-netische Beratung/strong bezeichnet, dient dazu, gene-tisch (mit-)bedingte Erkrankungen oder Risiken für Erkrankungen zu erkennen und zu verstehen. Sie wird von Fachärzten für Humangenetik durchge-führt, die eine spezielle Erfahrung mit erblichen Krankheiten haben, sowohl in der Diagnostik als auch in der Betreuung von betroffenen Personen und Familien. Eine humangenetische Beratung wird non-direktiv durchgeführt, es werden also keine Empfehlungen gegeben. Vielmehr sollen Hintergrün-de erklärt und Missverständnisse ausgeräumt wer-den, damit die Betroffenen in der Lage sind, selber die für sie richtigen Entscheidungen zu treffen./pp/ppEine humangenetische Beratung kann beispielsweise bei unerfülltem Kinderwunsch oder bei gehäuften Krebserkrankungen in der Familie hilfreich sein. Bei Vorliegen einer erblich bedingten Krankheit in einer Familie wird über deren Verlauf sowie über Präven-tionsmöglichkeite oder Behandlung aufgeklärt; bei Kinderwunsch kann über die Wahrscheinlichkeit des Auftretens einer genetisch bedingten Krankheit bei einem Kind gesprochen werden. Wenn bei einer Schwangerschaft beim heranreifenden Kind der Ver-dacht auf eine genetische Krankheit gestellt wurde, kann eine humangenetische Beratung dabei helfen, dass die Bedeutung der Befunde richtig verstanden wird und damit Entscheidungen vermieden werden, die später bereut werden könnten./pp/ppEine humangenetische Beratung sollte stets wertfrei sein. Wertende Begriffe wie „genetische Kompatibili-tät“ oder „defekte Gene“ werden im Rahmen einer humangenetischen Beratung vermieden./pp/ppGenetische Beratung ist in Deutschland eine Leis-tung aller gesetzlichen Krankenkassen und privaten Krankenversicherungen und wird daher von diesen bezahlt, wenn der Hausarzt oder ein Facharzt einen entsprechenden Überweisungsschein ausstellt./pp/ppemQuelle: /em/ppemWikipedia, Humangenetische Beratung, 20.06.2018/em/pp/pp/pp/pp/pp/pp/pp/pp/p
Stammbäume
pBei einer genetischen Beratung wird oft auch ein strongStammbaum /strongerstellt. Hierbei handelt es sich um eine grafische Darstellung der Mitglieder einer Fami-lie, wobei markiert wird, welche von ihnen Merk-malsträger der jeweils zur Frage stehenden Erb-krankheit waren oder sind. In einem solchen Stamm-baum stehen Kästchen für männliche und Kreise für weibliche Personen. Ein ausgefülltes Symbol kenn-zeichnet eine Person als Merkmalsträger; leere Sym-bole stehen für Personen, bei denen keine Sympto-me zu beobachten waren oder sind – die ausgefüll-ten bzw. leeren Symbole stellen somit den Phänotyp der Personen dar. In einem Stammbaum stehen die einzelnen Generationen untereinander: Die letzte Zeile zeigt die derzeit jüngste Generation. Durch Ver-bindungslinien wird verdeutlicht, wer mit wem Kin-der hervorgebracht hat (waagerechte Linie zwischen einer männlichen und einer weiblichen Person) bzw. wer das Kind welchen Paares ist (senkrechte Linien). Aufgrund eines Stammbaues kann in vielen Fällen der Genotyp der ratsuchenden Person(-en) abgelei-tet werden. Dies ermöglicht u. a. die Berechnung des Erkrankungsrisikos potentieller Kinder./p
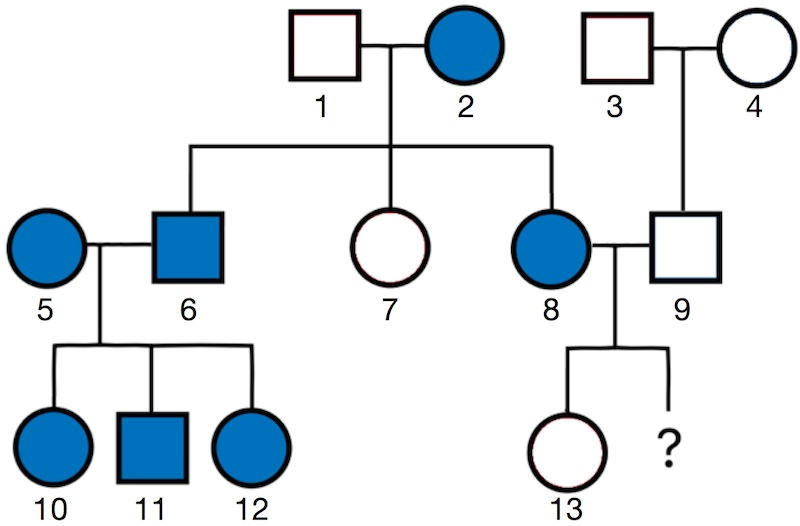
pDas Paar 89 erwägt ein weiteres Kind und hat die genetische Beratungsstelle aufgesucht, um sich zu den Risiken einer Erkrankung dieses Kindes beraten zu lassen. Die Frau (8) ist Betroffene, ihre Mutter (2) sowie ihr Bruder (6) ebenso. Ihr Vater (1) und ihre Schwester (7) sind gesund; auch ihr Mann (9) und seine Eltern (34) sowie das erste Kind des Paares (13) sind nicht betroffen, während ihre Schwägerin (5) und deren Kinder (10–12) Merkmalsträger sind./p


